CRISPR
CRIPSR (Clustered Regularly Interspaced Short Palindromic Repeats)
成簇规律间隔短回文重复序列,作为最新的基因组编辑工具,在生物学领域大放异彩。
常见的几种Cas蛋白Cas9、Cas12、Cas13和Cas14等,用于不同场景的CRISPR。
| CRIPSR蛋白 |
向导RNA |
PAM |
靶标 |
酶结构域 |
切割机制 |
| Cas9 |
sgRNA |
5’NGG |
DNA |
HNH,RuvC |
靶DNA中的特异性钝端双链断裂DSB |
| Cas12 |
crRNA |
5’TTN |
DNA |
RuvC-like |
在有5’overhangs的靶DNA中特异性DSB |
| Cas13a |
crRNA |
3’A、U、C |
RNA |
2X HEPN |
特异性RNA切割 |
| Cas13b |
crRNA |
/ |
RNA |
2X HEPN |
特异性RNA切割 |
| Cas14a |
sgRNA |
/ |
ssDNA |
RuvC-like |
在有5’overhangs的靶DNA中特异性DSB |
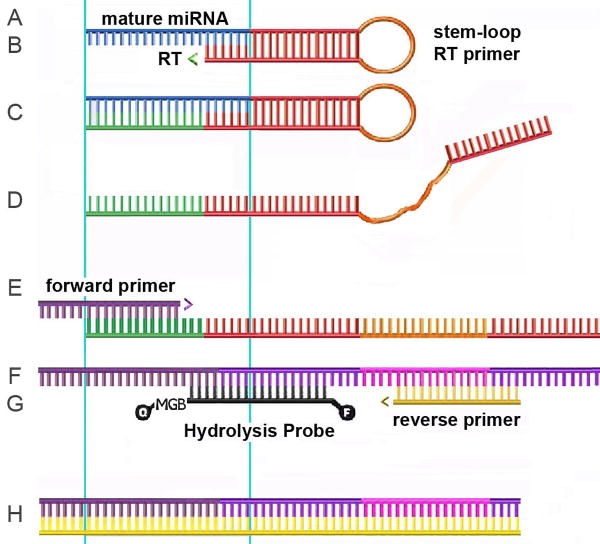
crRNA: CRISPR-derived RNAs,特异靶向DNA的RNA序列,自然界中存在。
tracrRNA: trans-activating RNA, 一般crRNA和tracrRNA互补配对形成双链,指导Cas9蛋白切割双链DNA。
sgRNA: single guideRNA 是人工制造的,自然界不存在。使用 CRISPR-Cas9 系统敲除基因表达或敲入特定突变时,
高质量的向导 RNAs (gRNAs) 的设计、产生和递送是成功的关键。
DSB: double-stranded DNA break 双链断裂。
RuvC: Cas9的两个结构域之一,RuvC结构域剪切非互补链。
HNH: Cas9的两个结构域之一,HNH核酸酶结构域剪切互补链。
HEPN: Higher Eukaryotes and Prokaryotes Nucleotide-binding domain, Cas13a含有两个HEPN结构域。
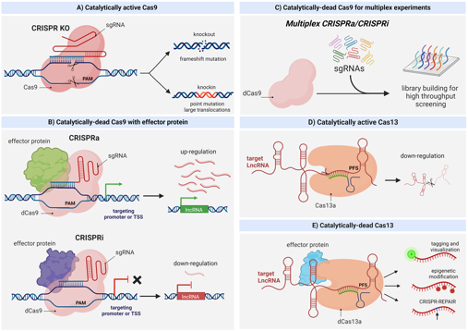
内源CRISPR途径被简化为两个主要组分:Cas9核酸酶和指导RNA(gRNA)。导向RNA是由crRNA和tracrRNA组成的双组分系统。crRNA靶向待切割的双链DNA,并且具有短的同源区域,允许其结合tracrRNA。 tracrRNA提供与Cas9蛋白结合的茎环结构。crRNA:tracrRNA双链体被称为gRNA。 Cas9核酸酶和gRNA形成Cas9核糖核蛋白(RNP),可以在整个基因组环境中结合并切割特定的DNA靶标。为了被RNP切割,靶必须具有两个特定序列。首先,gRNA需要17-21个碱基的RNA至DNA同源性,这被称为前间隔序列。 其次,Cas9蛋白需要有一个短的前间隔序列邻近基序(PAM),以结合靶DNA。如果存在连接tracrRNA,并且在gRNA和基因组靶标之间存在足够的同源性,则RNP切割靶DNA的两条链,在基因组中的该精确位置处产生DSB。
虽然细胞核中的功能性CRISPR是RNP形式,但作为分子工具的CRISPR的组件适合于各种递送方法。 早期实验成功地创建了嵌合单指导RNA或sgRNA,其将crRNA和tracrRNA组合成单个RNA链而不是天然存在的双链体。 该sgRNA和Cas9 mRNA可以从单个质粒表达,用于直接转染或包装成用于慢病毒转导的颗粒。 也可将重组Cas9蛋白与合成产生的crRNA和tracrRNA组合,以生成RNP转染或显微注射给胚胎。
靶向DSB形成后,细胞通常使用两种DNA修复途径中的一种来存活:非同源末端连接(NHEJ)或同源依赖性修复(HDR)。这些修复机制经常会出错,导致在靶位置诱变,或者通过破坏编码序列来功能性地失活或敲除基因(NHEJ),或者通过添加新的DNA序列来敲入特定的序列变化(HDR)。通过这种方式,CRISPR / Cas9系统能够对染色体DNA进行永久的、可遗传的修饰。此外,当Cas9核酸内切酶结构域失活并附着于其他效应分子以作用于基因组中的gRNA指定位点时,CRISPR系统可用作靶向递送系统。 这将CRISPR系统功能扩展到基因激活和抑制。最后,CRISPR系统可用于筛选
共表达的适当设计的gRNA指导Cas9切割靶序列,并在目的基因中产生DSB。然后可以通过三种方式中的任何一种实现基因敲除:1)细胞通过NHEJ修复断裂,导致切割基因的开放阅读框(ORF)内的随机插入或缺失(“插入缺失”);2)细胞通过HDR从用户提供的模板修复断裂,将特定的破坏性序列插入ORF中;3)一对gRNA产生两个DSB,其位于基本编码序列的侧翼,导致其切除。
靶向整合(基因敲入)通过HDR发生。为了通过HDR进行基因编辑,必须将含有所需序列的DNA “供体” 或修复模板与gRNA和Cas9一起递送至细胞,通常在供体质粒或寡核苷酸上。基因敲入的效率通常低于敲除(<10%的修饰等位基因),但可用于产生范围从单核苷酸变化至大插入物的特定修饰。
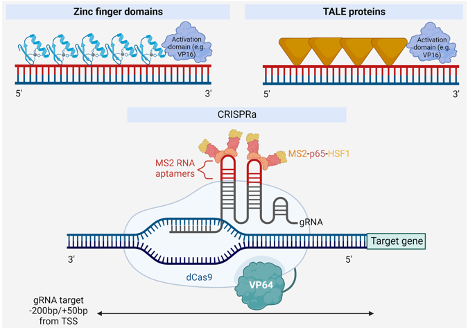
Cas9的一个独特特征是它能够独立于DNA切割而结合靶DNA,因为这是Cas9机制的两个独立步骤。野生型Cas9具有两个核酸酶结构域:RuvC和HNH。为了在没有切割的情况下实现结合,通过诱导点突变(SpCas9中的D10A和H840A)使两个核酸酶结构域失活,导致核酸酶死亡的Cas9(dCas9)。当与靶向转录起始位点的gRNA组合时,发现单独的dCas9足以通过阻断转录起始来降低或抑制转录。在这一发展之后,科学家开始尝试将转录抑制因子和激活因子与dCas9联系起来。
筛选可以快速调查大量候选基因或基因组位点,以参与您的途径或感兴趣的表型。池化寡核苷酸生产和大数据处理的改进,加上慢病毒递送的效率,使研究人员能够同时考虑数千个候选基因,无论是作为文库还是更为集中的子集(panel)。通常,子集(panel)和文库可以以两种形式组装:池化(在一个管中有数千个gRNA)或排列(在96孔板中每个孔一个gRNA)。
Summary of CRISPR/Cas tools
Cas9